microRNAs decoded. microRNAs may promote the reinnervation of the motor nerves by blocking the expression or synthesis of key substances. Video: Nature Publishing Group.
Virginia Tech’s Greg Valdez PhD knew that miR-206 was in the right place at the right time to help plug motor nerves back into muscles. The short non-coding RNA, discovered in 1994 by the late Washington University School of Medicine neuroscientist John Merlie PhD, is at the motor endplate and its synthesis is turned up upon detachment of the connecting nerve.
“That’s what led us to jump on this bandwagon,” says Valdez, “to try and find out what this microRNA is doing at the neuromuscular junction.”
His advisor, Harvard University’s Joshua Sanes PhD, got a phone call from University of Texas Southwestern’s Eric Olson PhD. He told him about graduate student Andrew Williams' latest results:
A (G93A-SOD1) mouse model of ALS without miR-206 appeared to progress much more quickly. And, died more than 3 weeks earlier due to the disease.
miR-206 might help stabilize neuromuscular junctions, keeping the motor nerves and muscles connected, thought Valdez.
He decided to take a closer look. He peered at their muscles under the microscope. Upon the onset of disease, more than half of their motor neurons were unplugged - more than double than those with miR-206 in their muscles.
miR-206 appeared to help rebuild neuromuscular junctions in ALS mice. And, protect them from the ravages of the disease.
Moving Target
In the earliest stages of ALS, most people feel their arms and legs gradually weaken. But their muscles keep moving – months after experiencing the first symptoms of disease.
The reason, according to a growing number of studies, is that many of the motor nerves reattach to muscles when they become unplugged. This 'collateral reinnervation' is one of many changes that occur in people with ALS to help protect them early in the disease course.
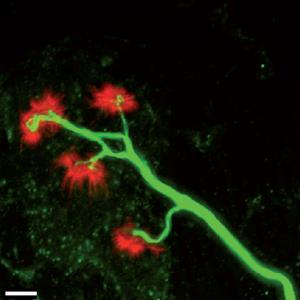
Compensatory sparks. Motor nerves plug back into muscles early in the disease course- keeping people with ALS moving. Image: Brunteau et al. Brain (2013). Courtesy of Oxford University Press.
By tapping into these compensatory mechanisms, clinicians hope to create medicines that help keep their patients with ALS moving longer by keeping the motor nerves and muscles connected.
But delivering miR-206 to people with ALS can be tricky to do. miR-206 must be specifically targeted to the neuromuscular junctions in people with ALS. And, must be packaged to protect the RNA from degrading en route.
Olson’s team is now working hard to figure out how miR-206 is turned up in people with ALS. The results might enable researchers to design treatments strategies that help keep people with ALS moving - by triggering miR-206 production before the muscles start to waste.
“We want this microRNA to come up as soon as there is any sign of neuromuscular destruction,” says Valdez. “That is the ultimate goal.”
Meanwhile, a growing number of researchers are trying to understand how miR-206 helps the motor nerves plug back into muscles in hopes to identify new treatment strategies for the disease.
To do that, scientists are working hard to identify proteins regulated by miRNA-206 in muscles. One enzyme, histone deactetylase 4 (HDAC4), an instigator of muscle atrophy, is emerging as a key target in its crosshairs.
HDAC4 production appears to be reduced by miR-206 in muscles according to results from Andrew Williams PhD, now at Columbia University. Introduction of miR-206 into cultured cells reduced enzyme levels about 60%. And, in mice lacking miR-206, levels of HDAC4 more than doubled in their muscles.
HDAC4 also appears to promote detachment of the motor nerves from muscles – at least in mice according to studies by Virginia Tech’s Greg Valdez PhD. Injured motor nerves appear to reattach more easily to muscles lacking HDAC4. Upon injury, nearly a third more motor neurons reconnected as compared to those in normal mice.
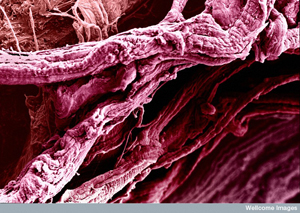
Fast and furious. HDAC4 appears to build up in fast twitch muscle fibers before disease onset - leading to muscle atrophy. Image: David Gregory and Debbie Marshall, Wellcome Images.
What’s more, HDAC4 appears to be turned up in people with ALS according to studies led by Duke University School of Medicine’s Tso-Pang Yao PhD. And, this increase appears to occur in key muscles affected by ALS before the onset of disease - at least in mice. Increased HDAC4 appears to be detected only in fast skeletal muscles of a G93A-SOD1 mouse model of ALS. The same muscles that ultimately fail in the disease.
The results suggest that miR-206 in part, helps motor neurons to reconnect in people with ALS early in the disease by lowering levels of HDAC4 in their muscles.
Reducing levels of HDAC4 might therefore be helpful in treating the disease.
***
Assistance Publique - Hôpitaux de Paris’ Pierre-François Pradat MD PhD suspected long ago that a lot could be learned from peering into the muscles of people with ALS. Key changes in muscles could contribute to ALS. And, exacerbate the disease.
These differences may enable clinicians to more accurately diagnose and monitor people with ALS. And, inspire new treatment strategies for the disease.
“Atrophy and weakness may not be simply collateral damage,” explains Pradat. “Crucial pathogenic events may occur in muscle fibers that could be reachable targets of future treatments.”
Pierre-François Pradat MD PhD got a call in 2000 from INSERM’s Jean-Philippe Loeffler PhD. He told him about Luc Dupuis PhDs latest results
Nogo-A builds up in the muscles of a (G86R-SOD1) mouse model of ALS before the onset of disease, said Loeffler.
Increased Nogo-A could prevent the motor nerves in people with ALS from being repaired and reconnected, Pradat thought.
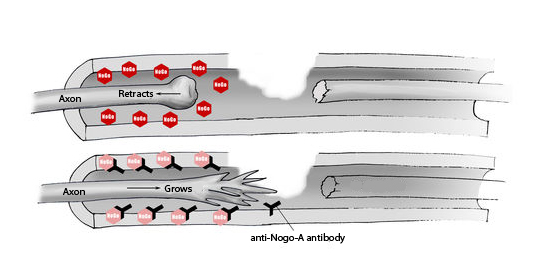
A go for anti-Nogo-A. Ozanezumab aims to help repair and reconnect injured motor axons by soaking up Nogo-A (red) in the surrounding debris. Learn more by tuning into our podcast with Pierre-François Pradat MD. Image(adapted): Uniklinik Bagrist, Switzerland.
Pierre-François Pradat MD PhD decided to look and see whether these same changes occurred in his patients with the disease.
He found that Nogo-A appeared to build up in the muscles of people with ALS. And, its levels appeared to correlate with the progression rate of the disease.
What’s more, people with high levels of Nogo-A suspected to have ALS are more likely to develop the disease. Nearly 90% of people who have detectable levels of Nogo-A in their weakening muscles develop tell tale signs of ALS including difficulties breathing, speaking and swallowing. Whereas more than 90% of people who appear to be Nogo-A ‘negative’ did not develop the disease.
The results suggested that reducing Nogo-A in people with ALS may help enable the motor nerves to reattach to muscles. And, slow progression of the disease.
An international phase II clinical trial of GlaxoSmithKline’s ozanezumab, a Nogo-A antibody that reduces Nogo-A levels, is ongoing.
"We hope that it could lead to a functional improvement and survival benefits in patients with ALS," says Pradat.
Now, Pierre-François Pradat MD PhD is using gene profiling to identify other changes in muscles in people with ALS in hopes to identify other potential strategies to treat and manage the disease.
His team reported in 2012 a fingerprint of 155 genes that appeared to reflect the degree of muscle decline in a small group of people with ALS. Expression profiles of 9 out of 155 genes could distinguish muscles that were seriously impaired.
One of the 9 genes was HDAC4.
The Sign of Four
ALS experts first looked to HDAC inhibitors in hopes to protect motor nerves in people with ALS. Researchers, however, have since discovered that these strategies could potentially provide more benefits - including help keep muscles healthy.
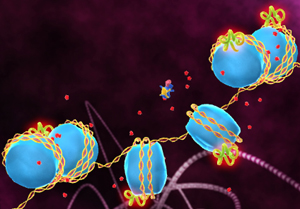
Tired muscle. HDAC4 turns off the expression of key genes by removing chemical marks (red) - leading to muscle atrophy. Image: National Cancer Institute.
HDAC4 promotes muscle atrophy – at least in mice. Increased levels of the enzyme induce muscle damage according to studies led by Duke University School of Medicine’s Tso-Pang Yao PhD. Whereas muscles without HDAC4 appear to resist atrophy – even after the motor nerves detach from them according to studies led by University of Texas Southwestern’s Eric Olson PhD.
What's more, increased levels of HDAC4 can be detected in muscles of people with ALS according to studies led by Duke University’s Tso-Pang Yao PhD.
But HDAC4 blockers might do much more for people with ALS then reduce muscle atrophy according to recent studies led by Assistance Publique - Hôpitaux de Paris’ Gaëlle Bruneteau MD. These strategies might help slow down the disease.
People who survived at least 5 years with ALS appeared to express at least one third less HDAC4 in their muscles than those that more rapidly declined. And, appeared to be better at keeping the muscles and motor nerves connected. Over half of the motor neurons appeared to plug back into the muscle fibers after becoming detached – more than double those in people with a more rapidly progressing form of the disease.
HDAC4 levels also appear to correlate with the rate of progression of the disease.
The results suggest according to Assistance Publique - Hôpitaux de Paris’ Pierre-Francois Pradat MD PhD that the key to keeping people with ALS moving is not the number of normal muscle-motor nerve connections. But the ability of their muscles to plug back in upon becoming detached.
Blocking HDAC4 in people with ALS therefore might boost the reinnervation capacity of the motor nerves- slowing the disease.
”Inhibiting histone deacetylase 4 appears to be a very promising therapeutic strategy,” says Pradat.
HDAC4, on target? Tempero Pharmaceutical's Class IIa HDAC blocker (above) might be a first step toward developing HDAC4-targeted medicines. But whether this is the best approach to treat ALS remains an open question. Image: Courtesy of Nature Publishing Group.
Now, chemists across the globe are working hard to cook up HDAC4-targeted medicines in hopes to create treatments for a number of diseases including ALS. In 2013, the GlaxoSmithKline spinoff Tempero Pharmaceuticals unveiled its Class IIa HDAC blockers which target HDAC4, HDAC5, HDAC7 and HDAC9 – a key first step in developing HDAC4-specific treatment strategies. The compounds are now being developed to treat autoimmune diseases including multiple sclerosis.
Meanwhile, Duke University School of Medicine’s Tso-Pang Yao PhD is taking a different approach to develop HDAC4-specific medicines. The potential therapies according to Yao might be helpful in treating many diseases neuromuscular in nature including ALS.
Whether targeting HDAC4 is the right strategy to treat ALS, however, remains an open question according to Virginia Tech's Greg Valdez PhD. HDAC4 plays a number of critical roles - including those outside skeletal muscles. The regulatory enzyme helps the heart keep up with demand by powering the remodeling of heart muscles. And, helps maintain vision by providing ‘life support’ to key nerves.
But according to Duke University School of Medicine’s Tso-Pang Yao PhD, these potential treatments may alleviate muscle weakness but leave other systems relatively unscathed. The reason according to Yao is that its enzymatic activity does not appear likely to be needed – at least to regulate heart load.
“In principle, this offers a therapeutic window where targeting HDAC4 catalytic activity would suppress atrophy but with limited toxicity,” explains Yao.
Nevertheless, miR-206 regulates the production of a number of substances in muscles according to Virginia Tech Greg Valdez PhD. Other key proteins may need to be downregulated in people with ALS to help the motor nerves reattach.
Now, Valdez’s team is working hard to understand how miR-206 helps protect neuromuscular junctions in people with ALS. The regulatory switches he identifies might form the basis of new treatments of the disease.
In future, he hopes to develop strategies to deliver miR-206 directly to people with ALS.
“In a disease like this, you have to consider everything,” says Valdez.
***
To learn more about emerging treatment strategies that aim to keep the muscles and motor nerves connected, tune into out podcast A Go for anti-Nogo-A. To find out more about HDAC-targeted treatment strategies amd their potential benefits for people with ALS, check out Emerging Potential of HDACIs in ALS.
References
Bruneteau, G. et al. (2013) Muscle histone deacetylase 4 upregulation in amyotrophic lateral sclerosis: potential role in reinnervation ability and disease progression. Brain 136, 2359-2368. Abstract | Full Text (Subscription Required)
Lobera, M. et al. (2013) Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nature Chemical Biology 9(5), 319-325. Abstract | Full Text (Subscription Required)
Choi, M.C., Cohen, T.J, Barrientos, T., Wang, B., Li, M., Simmons, B.J., Yang, J.S., Cox, G.A., Zhao, Y. and Yao T.P. (2012) A direct HDAC4-MAP kinase crosstalk activates muscle atrophy program. Molecular Cell 47(1), 122-132. Abstract| Full Text
Williams, A.H., Valdez, G., Moresi, V., Qi, X., McAnally, J., Elliott, J.L., Bassel-Duby, R., Sanes, J.R. and Olson, E.N. (2009) MicroRNA-206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science 326, 1549-1554. Abstract | Full Text (Subscription Required)
Cohen, T.J., Barrientos, T.,, Hartman, Z.C., Garvey, S.M., Cox, G.A., and Yao, T.P. (2009) The deacetylase HDAC4 controls myocyte enhancing factor-2-dependent structural gene expression in response to neural activity. FASEB Journal 23(1), 99-106. Abstract | Full Text
Cohen, T.J., Waddell, D.S., Barrientos, T., Lu, Z., Feng, G., Cox, G.A., Bodine, S.C. and Yao, T.P. (2007) The histone deacetylase HDAC4 connects neural activity to muscle transcriptional reprogramming. Journal of Biological Chemistry 282(46), 33752-33759. Abstract | Full Text
Learn more about GSK's ozanezumab
Pradat, P.F. et al. (2012) Muscle gene expression is a marker of amyotrophic lateral sclerosis severity. Neurodegenerative Diseases 9(1), 38-52. Abstract | Full Text (Subscription Required)
Pradat, P.F. et al. (2007) Muscle Nogo-A expression is a prognostic marker in lower motor neuron syndromes. Annals of Neurology62(1), 15-20. Abstract | Full Text (Subscription Required)
Jokic, N., Gonzalez de Aguilar, J.L., Dimou, L., Lin, S., Fergani, A., Ruegg, M.A., Schwab, M.E., Dupuis, L. and Loeffler, J.P. (2006) The neurite outgrowth inhibitor Nogo-A promotes denervation in an amyotrophic lateral sclerosis model. EMBO Reports 7(11), 1162-1167. Abstract | Full Text
Jokic, N. et al. (2005) Nogo expression in muscle correlates with amyotrophic lateral sclerosis severity. Annals of Neurology 57(4), 553-556. Abstract | Full Text (Subscription Required)
Learn more about the neuromuscular junctions
Burden, S.J. (2011) SnapShot: Neuromuscular Junction. Cell 144(5), 826-826.e1. Illustration (Subscription Required)