Since the discovery of the first ALS-associated gene, superoxide dismutase 1 (SOD1) in 1993, scientists tested more than 30 experimental ALS drugs in clinical trials. Only Sanofi’s Rilutek is FDA approved and at best moderately treats the disease.
A big part of the problem, says ALS experts, is that the disease is extremely variable and the underlying mechanism of the disease is not understood. But despite these obstacles, University of Torino scientists argue based on findings of a new study that more can be done now to increase the chances for a new drug to be successfully developed simply by selecting patients that participate in clinical trials differently.
”I think what we have shown,” says lead author Adriano Chiò MD, “indicates that we are making mistakes. It is time to correct the mistakes.”
The study is published this month in the journal Neurology.
The University of Torino team compared the local ALS patient population that participated in clinical trials to those diagnosed with the disease between 2003 and 2008. The researchers found that those enrolled in clinical trials tended to be younger, male and half as likely to have the bulbar form of the disease.
These results suggest that experimental ALS medicines may be more difficult to demonstrate to be effective because the people participating in clinical trials tend to be healthier and have a more slowly progressing form of the disease.
The University of Torino team suggests clinical trial investigators should accommodate a broad range of people with ALS by dropping forced vital capacity minimums to 60%. And these researchers say that clinical trials should instead include people with possible ALS, people with probable ALS or people who are recently diagnosed with the disease. The researchers argue that by sticking to these patients, the interventions also have a much better chance to be effective since these people are in the early stages of the disease.
“The inclusion of patients in the earliest phase of the disease, when more motor neurons are still present, increase the probability that a drug may be demonstrated to be effective,” explains Chiò.
Rethink the possible
Many of these recommendations have already been put in place. Within the last two years, clinical trials started to include broader populations of people with ALS by reducing their forced vital capacities to as low as 50%. And researchers are opening their doors to people suspected to have the disease. Ongoing trials such as Biogen Idec’s Empower (dexpramipexole), Cytokinetics’ CK-2017357 and GlaxoSmithKline’s “NOGO” trials include people with only a possible ALS diagnosis.
“There has been significant changes in how the trials are designed,” says Massachusetts General Hospital ALS Clinic Director Merit Cudkowicz MD, MSc. “We already recognize that inclusion criteria were too strict.”
The reason drug developers feel comfortable with including people with possible ALS says Director of Duke University’s ALS Clinic, Rick Bedlack MD, PhD, MS, is that there is growing evidence that these people are extremely like to get the disease. “Once you get to the category of possible ALS the chances that you are going to move into one of those other areas is incredibly high,” says Bedlack.
And says Knopp Biosciences’ Valentin Gribkoff, who heads the dexpramipexole team, opening the door to people in earlier stages gives them the best chance of detecting efficacy.
“What we think that did for us is allow for a wider-dynamic range for seeing a drug effect,” explains Gribkoff.
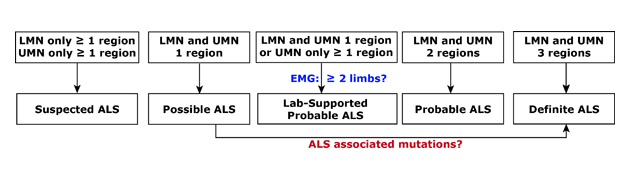
Diagnosis 101. ALS is often diagnosed in stages which are defined by the number and location of areas affected and the degree of disease spread. Called El Escorial criteria, a combination of clinical, electrical, and imaging measures are typically used. LMN, lower motor neurons. UMN, upper motor neurons. EMG, electomyography.
Reality Check
While more and more clinical trials open their doors to people with the first signs of ALS, some researchers however continue to keep them open to prevalent cases – people up to two years into ALS - even when an intervention is expected to be most effective early in the disease.
One reason says Cudkowicz is that the diagnosis of definite ALS with today’s tools typically takes 12 to 14 months. It is just not possible to test interventions in people with definite ALS, at least in the U.S., within the first year of experiencing symptoms of the disease.
But this is not the only reason researchers are hesitant to focus clinical trials on so-called incident cases of the disease. With only a small percentage of people participating in clinical trials, some ALS experts worry that these restrictions will make it that much harder to develop promising medicines for the disease.
“We are really going to magnify the enrollment problem,” says Bedlack. “It’s probably going to be even slower and more difficult to recruit patients.”
Chiò disagrees. He says that testing early interventions under these restrictions is doable – particularly in large cities in which 100s of cases of ALS are diagnosed annually– and is necessary to give the drugs their best chance of being demonstrated to be effective.
“I hope that trials will be modified and will take into account the problems I have found,” says Chiò.
Trials and Tribulations
With the prospect of a growing number of trials however, being restricted to people who experience their first foot drop within 24 months, people with ALS are understandably frustrated. But says Cudkowicz this is not going to be the case for each and every drug being developed for the disease. Such decisions are made case by case based on when the medicines are most likely to work during the course of the disease. As researchers learn more about ALS, new treatment strategies may emerge that target more advanced stages of ALS - treatment strategies that must be evaluated in those populations.
“I don’t think there is one model fits all clinical trials,” says Cudkowicz.
References
Chiò, A., Canosa, A., Gallo, S., Cammarosano, S., Moglia, C., Fuda, G., Calvo. A, and Gabriele, M.. (2011) ALS clinical trials: Do enrolled patients accurately represent the ALS population? Neurology, 77(15), 1432-1437.
Further Reading
Beghli, E. et al. (2011). The epidemiology and treatment of ALS: focus on the heterogeneity of the disease and critical appraisal of therapeutic trials. Amyotrophic Lateral Sclerosis, 12(1), 1-10.
Bedlack, R.S., Wicks, P., Heywood, J. and Kasarskis E. (2010). Modifiable barriers to enrollment in American ALS research studies. Amyotrophic Lateral Sclerosis, 11(6), 502-507.
UPDATED 11/21/11: Knopp Biosciences' Val Gribkoff joins the conversation.