With the discovery of nearly 20 genes linked to ALS, researchers are unraveling this complex disease at increasing speed. But, according to recent research, there is much more than a handful of genetic errors in people with ALS that contributes to the destruction of the motor nerves.
The epigenome – the chemical tags that mark which genes are turned on and off in our tissues – is emerging as a new player in neurodegenerative diseases including ALS. Inherently flexible, the epigenome rapidly evolves to help people adapt to a crash diet, stress or simply a change in scene. But in people with ALS, studies suggest, these changes might actually fuel the progression of the disease.
Emerging drugs that restore or remove these so-called epigenetic marks may have the potential to treat ALS. But figuring out how to safely reset these chemical switches in motor neurons using these medicines, according to experts, may be tricky.
AcE AcE Baby
Critical genes are switched on or off in specific tissues in part by adjusting their accessibility to the intracellular decoding machines. Known as acetylation, chromatin packing proteins called histones get pushed aside, allowing these genes to be transcribed and proteins, produced. But in people with a growing number of neurological conditions including ALS, the levels of histone acetylation drops, contributing to neurodegeneration.
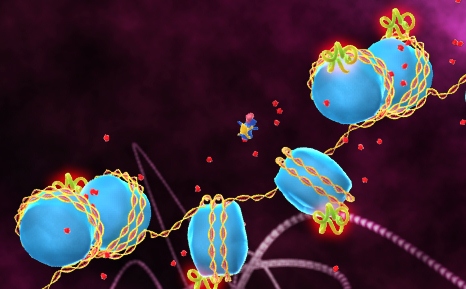
HDACs Crash. Histone deacetylases (green) remove acetyl groups (red) rendering the genes unreadable by the transcriptional machinery (dark blue). Image: National Cancer Institute.
Epigenetics first grabbed neuroscientists’ attention in 2001 when University of California Irvine scientists Leslie Thompson PhD and Larry Marsh PhD reported that the loss of histone acetylation in fruit flies contributed to Huntington’s disease. The team found that the fruit flies did not produce a critical histone acetyltransferase (HAT). But treatment with histone deacetylase enzymes (HDACs) blockers to boost acetylation levels stopped the disease in its tracks.
“These results really created enormous awareness,” explained New York Burke Rehabilitation Center neurologist Raj Ratan MD, “that small molecule [HDAC] inhibitors that had been developed for cancer might actually have benefit for neurodegenerative diseases.”
Subsequent preclinical studies found that several compounds that broadly block HDACs appeared to slow neurodegeneration. But researchers still remain unsure why exactly these drugs might be therapeutically beneficial.
Scientists speculate that HDAC inhibitors may switch on the production of substances that helps nerves withstand stresses associated with these diseases. A multi-institutional team led by Raj Ratan, for example, found in 2003 that the HDAC inhibitor trichostatin A increased the levels of acetylated transcription factor Sp1 in neurons, boosting the production of proteins that help protect them from the flood of damaging reactive oxygen species (ROS). And in 2006 and 2008, researchers from the National Institutes of Health in Washington D.C. found that HDAC inhibitors sodium phenylbutyrate, trichostatin A and valproic acid encouraged astrocytes to produce so-called neurotrophins that protected cultured neurons from destruction.
Lost In Translation
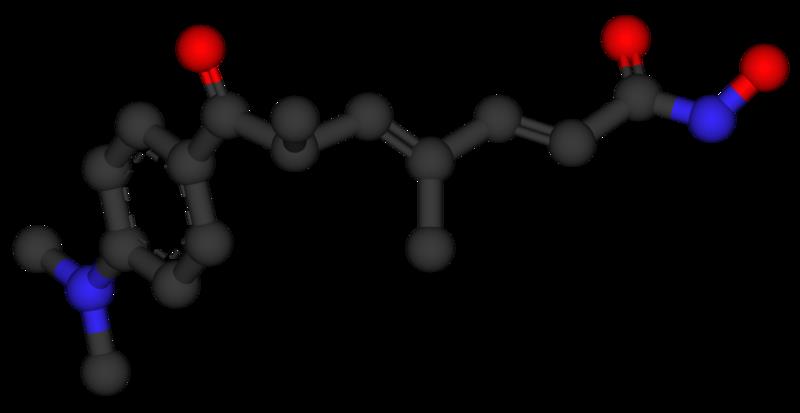
Trichostatin A. Researchers from University of Southern California reported in 2011 that trichostatin A could reduce motor neuron loss and extend survival of mice experiencing symptoms similar to ALS.
Researchers first began to suspect that epigenetic changes contributed to ALS in 2003 when scientists reported that levels of a critical histone deacetlyase in mice dropped 70% primarily in degenerating motor nerves. Subsequent preclinical studies indicated that three broad spectrum histone deacetylase inhibitors – sodium phenylbutyrate, trichostatin A, and valproic acid – might be useful in treating ALS because treatment delayed clinical onset and/or extended survival of mouse models. But independent research teams at ALS TDI in Massachusetts were unable to reproduce these findings for any of these medicines. And, a phase III clinical trial of 160 ALS patients in the Netherlands found that valproic acid at least at a dose typically prescribed for epilepsy, did not appear beneficial.
A critical limitation of today’s HDAC blockers according to some experts is these drugs may simply need to be administered at too high a dose to be effective.
An estimated 3-5% of genes in people are switched on by today’s histone deacetylase inhibitors such as trichostatin A according to experts. Many of these genes protect the motor nerves from further deterioration. But many others likely promote their destruction.
Researchers are now working hard to develop potential medicines that block specific “classes” of histone deacetylases in hopes to identify a drug that more specifically switches on neuroprotective agents to improve the safety and tolerability of these medicines. These include Repligen’s Class I HDAC1, HDAC2, HDAC3 and HDAC8 inhibitor RG2833 which is being developed for the treatment of Friedrich’s ataxia and GlaxoSmithKline spinoff Tempero Pharmaceuticals’ Class IIa HDAC4, HDAC5, HDAC7 and HDAC9 inhibitors, for the treatment of chronic inflammatory diseases including potentially multiple sclerosis.
Meanwhile, researchers elsewhere are going for the gold hoping to create potential medicines that selectively block specific HDAC enzymes to treat neurological diseases. MIT neuroscientist Li-Hui Tsai PhD, for example, hopes to create potential medicines for Alzheimer’s disease by generating compounds that specifically block HDAC2 to treat memory loss.
”I think you are really seeing the HDAC inhibitor [field] blossom,” says Ratan.
The jury is still out however whether any of these more selective drugs being developed are therapeutically beneficial in ALS.
Despicable ME
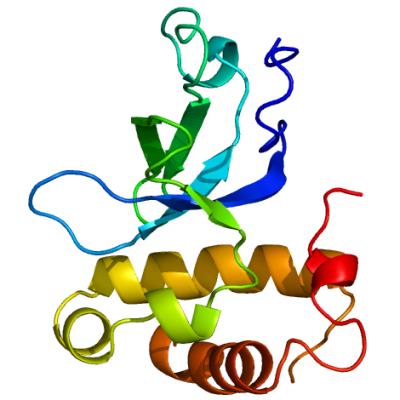
All about ME. Researchers discovered that the DNA methyltransferase Dnmt3A may trigger the deterioration of the motor nerves. Image: Structural Genomics Consortium, University of Toronto.
Johns Hopkins neuroscientist Lee Martin PhD however, suspected that there was much more than changes in histone acetylation that occur in people with ALS. Critical genes could also be inactivated by methylation in motor neurons by DNA methyltransferases, triggering the destruction of the motor nerves. Enzymes therefore, that could also be targeted to treat the disease.
“Seeing that there was promise in histone modifications,” explained Martin, “maybe there could also be some promise in [targeting] DNA methylation.”
To determine whether increases in methylation could also lead to ALS, Martin’s team in the late 2000s measured the amounts of DNA methyltransferases in the brain and spinal cord of patients and healthy people. The researchers found that the levels of the two of these enzymes, Dnmt1 and Dnmt3A were significantly higher in the CNS of people with ALS, suggesting that hypermethylation of certain genes could indeed be contributing to the disease.
“There was no data to say that DNA demethylation,” however, explained Martin, “could actually cause neurodegeneration.”
So, the Johns Hopkins researchers turned to model systems. In culture, the researchers found that increased levels of Dnmt3A triggered the degeneration of mouse spinal cord motor neurons. And in mice, treatment with the DNA methyltransferase blocker RG108 protected the motor nerves nearly 100% after sustaining injury.
“It is an exciting first step,“ says Ratan. “I think this study has really implicated DNA methyltransferases in neurodegeneration in a way that no one has come close to."
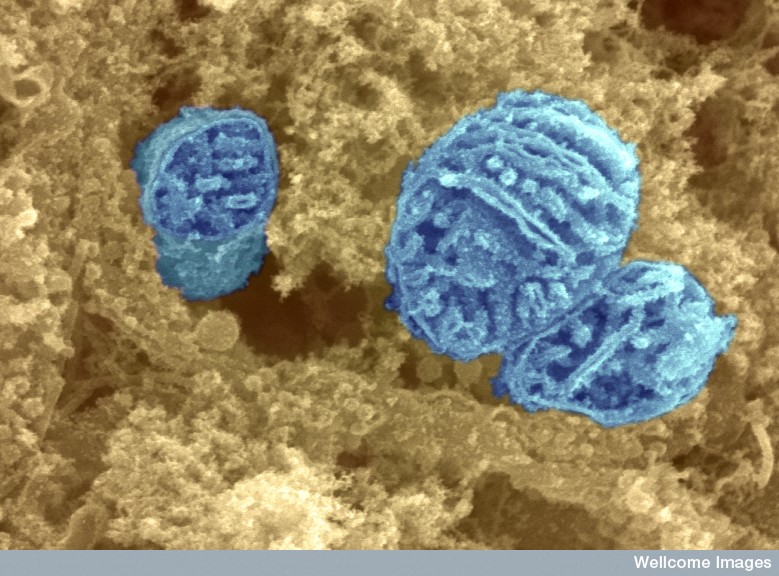
Circuit breaker. Power plants called mitochondria (above) may breakdown in motor neurons in people with ALS due to the hypermethylation of critical genes. Image: David Furness PhD, Wellcome Trust.
Intriguingly, the Johns Hopkins team found that one of these DNA methyltransferases, Dnmt3A, was present in the right place – the nerve terminals – to potentially trigger ALS. And, these enzymes could be found in these distal regions in mitochondria - potentially resulting in power outages, synaptic dieback and ultimately, the muscles to become unplugged.
“If we could actually show that ALS starts in the mitochondrial genomes at the nerve terminal,” says Martin, “that would be a new way of thinking.”
Now, Martin’s team is working hard to track down the genes in people with ALS that are silenced. Identifying these hypermethylated genes can give researchers a better idea of what signals could target the motor nerves for destruction, facilitating the design of better strategies to protect them in people with ALS.
But there may be no need to wait that long. Several pre-cancer or cancer drugs that target methyltransferases are being developed or are already in the clinic. Emerging medicines that could be evaluated for the ability to slow or stop ALS.
The jury is still out whether such methylation-reducing medicines could be safe and selective enough to be useful to treat a neurodegenerative disease such as ALS. But compared to potential medicines that target the histone deacetylases, there are considerable advantages to developing such a strategy for ALS according to Martin.
“If the changes occur at the nerve terminals at the neuromuscular junctions,” explains Martin, “we don’t have to worry about [the drug] crossing the blood brain barrier. That could not only be a new target but a new way of thinking about treating the disease.”
References
Chestnut, B.A., Chang, Q., Price, A., Lesuisse, C., Wong, M. and Martin, L.J. (2011). Epigenetic regulation of motor neuron cell death through DNA methylation. Journal of Neuroscience 31(46), 16619-16636. Abstract | Full Text (Subscription Required)
Del Signore, S.J., Amante, D.J., Kim, J., Stack, E.C., Goodrich, S., Cormier, K., Smith, K., Cudkowicz, M.E. and Ferrante R.J. (2009) Combined riluzole and sodium phenylbutyrate therapy in transgenic amyotrophic lateral sclerosis mice. Amyotrophic Lateral Sclerosis 10(2), 85-94. Abstract | Full Text
Piepers, S.,et al.(2009) Randomized sequential trial of valproic acid in amyotrophic lateral sclerosis. Annals of Neurology 66(2), 227-234. Abstract | Full Text (Subscription Required)
Rouaux, C., Panteleeva, I., René, F., Gonzalez de Aguilar, J.L., Echaniz-Laguna, A., Dupuis, L., Menger, Y., Boutillier, A.L. and Loeffler, J.P. (2007) Sodium valproate exerts neuroprotective effects in vivo through CREB-binding protein-dependent mechanisms but does not improve survival in an amyotrophic lateral sclerosis mouse model. Journal of Neuroscience 27(21), 5535-5545. Abstract | Full Text
Ryu H et al. (2003) Histone deacetylase inhibitors prevent oxidative neuronal death independent of expanded polyglutamine repeats via an Sp1-dependent pathway. Proceedings of the National Academy of Sciences 100(7), 4281–4286. Abstract | Full Text
Ryu, H et al. (2005) Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. Journal of Neurochemistry 93(5), 1087-1098. Abstract | Full Text (Subscription Required)
Scott, S. et al. (2008) Design, power, and interpretation of studies in the standard murine model of ALS. Amyotrophic Lateral Sclerosis 9(1), 4-15. Abstract | Full Text (Subscription Required)
Steffan JS et al. (2001) Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature 413(6857), 739–743. Abstract | Full Text (Subscription Required)
Sugai F, Yamamoto Y, Miyaguchi K, Zhou Z, Sumi H, Hamasaki T, Goto M, Sakoda S. (2004) Benefit of valproic acid in suppressing disease progression of ALS model mice. European Journal of Neuroscience 20(11), 3179-3183. Abstract | Full Text (Subscription Required)
Van Lint, C., Emiliani, S. and Verdin E (1996). The expression of a small fraction of cellular genes is changed in response to histone hyperacetylation. Gene Expression 5(4–5), 245–253. Abstract | Full Text (Not available online)
Wu X et al. (2008) Histone deacetylase inhibitors up-regulate astrocyte GDNF and BDNF gene transcription and protect dopaminergic neurons. International Journal of Neuropsychopharmacology 11(8):1123–1134. Abstract | Full Text
Yoo Y.E. and Ko C.P. (2011) Treatment with trichostatin A initiated after disease onset delays disease progression and increases survival in a mouse model of amyotrophic lateral sclerosis. Experimental Neurology 231(1), 147-159. Abstract | Full Text (Subscription Required)
Further Reading
Fischer, A., Sananbenesi, F., Mungenast, A. and Tsai, L.H.. (2010). Targeting the correct HDAC(s) to treat cognitive disorders. Trends in Pharmacological Sciences 31(12), 605-617. Abstract | Full Text
Sleiman, S.F., Basso, M., Mahishi, L., Kozikowski, A.P., Donohoe, M.E., Langley, B. and Ratan, R.R. (2009). Putting the 'HAT' back on survival signalling: the promises and challenges of HDAC inhibition in the treatment of neurological conditions. Expert Opinion Investigational Drugs 18(5), 573-584. Abstract | Full Text
Urdinguio, R.G., Sanchez-Mut, J.V. and Esteller, M (2009). Epigenetic mechanisms in neurological diseases: genes, syndromes, and therapies. Lancet Neurology 8(11), 1056-1072. Abstract | Full Text (Subscription Required)
Learn more about epigenetics and disease
Watch Epigenetics on PBS. See more from NOVA scienceNOW.