Mousehunt Researchers are working hard to create mouse models of ALS to identify and evaluate potential medicines for the disease. Image: Rama, Wikimedia.
More than 30 potential therapies for ALS have been tested in the clinic. But only riluzole is FDA-approved to treat the disease. And, boosts survival of people with ALS just 2 to 3 months.
In hopes to change that, researchers are working hard to develop new mouse models of ALS to help identify and prioritize potential therapies to push forward into the clinic.
Now, an international research team led by Cardiff University neuroscientist Natalia Ninkina PhD, introduce a new mouse model of ALS. The transgenic mouse, which produces a truncated form of the protein Fused In Sarcoma (FUS), exhibits key signs of ALS including progressive muscle atrophy and paralysis. And, accumulates key delocalized proteins in the brain and spinal cord.
The mouse is the first FUS-linked model of ALS to be published that exhibits key signs of ALS and key histological features of the disease.
The study is published this month in the Journal of Biological Chemistry.
In 1994, Northwestern University’s Mark Gurney PhD introduced the first mouse model of ALS in hopes to develop treatments for the disease. The transgenic mouse, which harbors an ALS-associated mutation in the superoxide dismutase 1 (SOD1) gene, exhibits key signs of ALS including muscle decline and paralysis. And, dies due to respiratory failure.
Nearly two decades later, however, riluzole remains the only drug identified to date that benefits people with ALS. One reason, argues some scientists, is that ALS is an extremely heterogeneous disease. More than 20 genes by some estimates appear to be linked to familial ALS. And, the survival of people with ALS is extremely variable – just months to more than a decade.
Multiple mouse models of ALS may be needed to identify treatments that benefit people with such a heterogeneous disease.
An RNA world?
In the mid-2000s, researchers turned their attention to TDP-43, a protein that appears to buildup in the brain and spinal cord of more than 90% of cases of ALS. What’s more, TDP-43 appears to be altered in about 1 in 20 cases of inherited form of the disease.
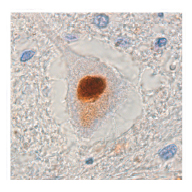
A big FUS? Many people with ALS accumulate FUS in the cytoplasm of motor neurons, suggesting that its deregulation may contribute to the disease. Image courtesy of Nature Publishing Group.
But the TDP-43 mouse models of ALS published to date do not appear to fully recapitulate the disease. Progressive motor neuron loss could be detected. And, walking appears to be affected. But death appears to be due to defects in the gut – uncharacteristic of the disease.
A growing group of research teams are now setting their sights on the transcription factor FUS in hopes to develop additional mouse models of the disease. The protein, like TDP-43, appears to be altered in about 1 out of 20 cases of familial ALS. And, appears to aggregate in the cytoplasm of motor neurons in many more cases of ALS – including sporadic disease.
Now, researchers at Cardiff University in Wales introduce a new FUS model of ALS. The transgenic mice, which produce a truncated form of the protein, exhibit key symptoms of ALS including progressive muscle weakness – often beginning in a single limb. And, rapidly develop paralysis.
Taking a look under the microscope, the motor neurons of these mice appear to sustain extensive damage due to inflammation leading to the crumbling of the neuromuscular junctions – much the same as in people with the disease.
The jury, however, is still out whether these mice can be used to develop drugs for ALS. The onset of disease is extremely variable. The first signs of muscle weakness occur at any time during a 2 month period – as compared to about 2 weeks in the G93A SOD1 mouse model of the disease.
The mice may nevertheless help prioritize potential ALS therapies to advance to the clinic by helping to validate them.
In the meantime, a US team led by University of California San Francisco’s Eric Huang MD PhD is taking a more conventional approach to develop a FUS model of ALS. The transgenic mice, which harbors the R521C ALS-associated mutation in the FUS gene, display key signs of ALS including spasticity, synaptic loss and motor decline according to results presented at SfN 2012. And, exhibit certain histological hallmarks of disease including delocalized FUS in the spinal cord. Analysis is ongoing.
***
To learn more about emerging mouse models of ALS, check out SfN12: ALS, Down on the Bayou. To find out about stem cell models of ALS and their potential to discover medicines for the disease, check out iPS ready, set, screen.
References
Shelkovnikova TA et al. (2013) Fused in Sarcoma (FUS) Protein Lacking Nuclear Localization Signal (NLS) and Major RNA Binding Motifs Triggers Proteinopathy and Severe Motor Phenotype in Transgenic Mice. Journal of Biological Chemistry, doi: 10.1074/jbc.M113.492017 Abstract | Full Text
Mitchell, J.C. et al. (2013) Overexpression of human wild-type FUS causes progressive motor neuron degeneration in an age- and dose-dependent fashion. Acta Neuropathologica 125(2), 273-288. Abstract | Full Text
Huang, C., Zhou, H., Tong, J., Chen, H., Liu, Y.J., Wang, D., Wei, X. and Xia, XG. (2011) FUS transgenic rats develop the phenotypes of amyotrophic lateral sclerosis and frontotemporal lobar degeneration. PLoS Genetics 7(3), e1002011. Abstract | Full Text
Kwiatkowski, T.J. et al. (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323(5918), 1205-1208. Abstract | Full Text (Subscription Required)
Vance, C. et al. (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323(5918), 1208-1211. Abstract | Full Text (Subscription Required)
Further reading
McGoldrick, P., Joyce, P.I., Fisher, E.M., Greensmith, L. (2013) Rodent models of amyotrophic lateral sclerosis. Biochimica Biophysica Acta 1832(9), 1421-36. Abstract | Full Text (Subscription Required)