Amyotrophic Lateral Sclerosis (ALS) is the most common among various disorders affecting motor neurons. Many other motor neuron disorders show signs and symptoms that often overlap with those that manifest in people living with ALS. Further, the biological processes implicated as disease drivers in ALS may also play roles in these other diseases. These areas of ambiguity can lead to confusion for people living with, treating, and developing new treatments for these disorders.
Primary lateral sclerosis (PLS) and progressive muscular atrophy (PMA) are arguably most closely related to ALS clinical manifestation, albeit in very different ways. Other related disorders include frontotemporal dementia (FTD) and chronic traumatic encephalopathy (CTE) with symptoms largely divergent from those observed in people living with ALS, but with overlapping genetic risk factors and similar pathological hallmarks in central nervous system tissues.
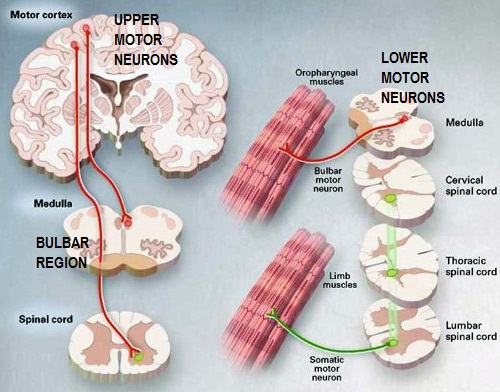
Motor neurons that connect to, or innervate, skeletal muscles directly from the brainstem and spinal cord are called lower motor neurons. The motor neurons that connect from the motor cortex in the brain to the lower motor neurons in the spinal cord are called upper motor neurons. Motor neuron diseases are caused by the dysfunction of these cells, with some forms being defined by the involvement of either or both sets of motor neurons.
Specific muscles are stimulated by specific populations of upper and lower motor neurons in the brain and spinal cord. That means that signs and symptoms can be mapped to general areas of the affected person’s brain and spinal cord. The type and location of symptoms at a person’s disease onset, coupled with observations of whether or how those signs and symptoms spread and worsen across different muscles largely defines which motor neuron disease a person will be diagnosed with.
(Image Source: Emory University Website)
Motor neuron diseases are driven by biological pathways that are still being unraveled by scientists. While there are infantile, juvenile, and adult onset motor neuron disease types, this report aims to draw distinctions between adult onset disorders including the aforementioned ALS, PLS, PMA, and progressive bulbar palsy (PBP).
Is it ALS or not?
No two cases of motor neuron disease are exactly the same. Some people report initial symptoms such as muscle weakness in their hands, others report a general clumsiness on their feet or foot drop, while still others experience a slurring of speech. In isolation, there are many possible explanations for any of those symptoms which do not include motor neuron diseases. Ultimately, it is the progressive worsening of symptoms in a person over time that leads to a diagnosis of a motor neuron disease.
As a result of the need to see a progressive loss of function, the ALS diagnostic process is often described as diagnosis by exclusion of other potential diseases along the way. That is to say that many other possibilities are ruled out before arriving at the final diagnosis of ALS.
A scale, called the El Escorial Criteria, was developed to help standardize the process by which a physician can determine whether or not a person has ALS. The El Escorial Criteria calls for the assessment of upper and lower motor neurons in four distinct body quadrants roughly described as: head/neck, arms/shoulders, trunk, and legs. Depending on which areas of the central nervous system are being impacted, a person may receive a suspected, possible, probable or definite diagnosis of ALS.
A person must show progressive loss of function in both the upper and lower motor neurons to be diagnosed with ALS. Whether or not that is occurring can be determined using a series of clinical tests, including electromyograms (EMG), nerve conduction studies, and magnetic resonance imaging (MRI). In addition, some neurologists increasingly use assessments of cerebral spinal fluid, blood, and muscle biopsies to solidify diagnoses. While these and other tests are part of the process, not everyone’s experience will be the same. Some may be given a diagnosis in a matter of days while others report it taking a year or longer to be diagnosed with ALS.
However, it is not uncommon for a person to be given a diagnosis of “suspected” or “possible” ALS and then after several months of little or no discernable progression for the diagnosis to be modified to a different form of motor neuron disease such as PLS, PMA or PBP, or a completely different disorder all together. In this sense, it is not uncommon for a person to be given more than one diagnosis within the spectrum of motor neuron diseases over time depending on their specific case and progression profile.
Lower Motor Neuron Disorders
Progressive bulbar palsy (PBP) is a form of motor neuron disease that mostly affects the face, throat and tongue muscles. Research suggests that PBP impacts the lower motor neurons to a greater extent than upper motor neurons. PBP will make it hard and eventually impossible for a person to speak or swallow on their own. Sometimes PBP is called bulbar onset motor neuron disease because of the unique way in which it manifests itself. Because PBP affects the muscles in a person’s mouth so acutely, they are more at risk to inhale saliva or food particles that can cause respiratory infections and other issues. PBP is a comparatively aggressive from of motor neuron disease. While there is some debate over whether PBP should be classified as a kind of ALS, most ALS specialists do classify it as such.
Another form of motor neuron disease is called progressive muscular atrophy (PMA), which has been found to only impact the lower motor neurons. Sometimes people with PMA experience symptoms limited to on their arms or legs. Due to the significantly focal atrophy across a person’s limb muscles, a doctor may also call this subtype by other names, such as “flail arm” or “flail leg” disorder. In general, people diagnosed with PMA have a better prognosis than other forms of motor neuron disorders with survival being twice as long, on average, than those diagnosed with ALS.
Primary Lateral Sclerosis
On the other end of the spectrum of motor neuron diseases is primary lateral sclerosis (PLS), a disease in which dysfunction is limited to only the upper motor neurons. Those diagnosed with PLS typically experience weakness in their legs or hands, with more rare cases of PLS starting in a person’s tongue. When seeking a diagnosis, one test typically conducted on the patient along the way is an EMG. Should that test come back ‘normal’, it can indicate that loss of function in the lower motor neurons can be ruled out at that time and a physician may give a diagnosis of PLS. As with PMA, a person with PLS is expected to survive for many years following initial onset of symptoms. However, in some cases, those initially diagnosed with PLS are diagnosed later with ALS if they develop signs of loss in other regions specifically those associated with lower motor neurons.
What about FTD and CTE?
Some forms of frontotemporal dementia (FTD) have shown evidence of occurring together with ALS. Specifically, FTD has been associated with ALS cases linked to mutations in TDP-43, C9orf72, FUS, and VCP. FTD is a disease in which the frontal and temporal lobes of the brain experience atrophy and neuronal death. This pathology causes progressive dementia which can manifest in changes in a person’s behavior, cognitive or social abilities over time. While less than half of those diagnosed with ALS will experience some form of dementia or other cognitive, social or behavioral issue during their disease course, regular cognitive testing has become part of standard ALS care, enabling earlier interventions to be applied. Not all of those with ALS that experience some form of dementia will be diagnosed with FTD, which makes a duel diagnosis of ALS/FTD relatively rare.
Another neurodegenerative disease often associated by the media and general public with ALS is chronic traumatic encephalopathy (CTE). This is a disease that is found mostly in individuals that have experience several injuries to the brain, causing significant brain damage. While CTE has been associated in popular culture with contact sports, such as American football, boxing and soccer, it is not known just how much head trauma a person must experience for them to develop CTE. Those suffering from CTE can experience symptoms similar to ALS, including progressive muscle weakness, atrophy and generalized loss of ability. Some may call CTE an “ALS mimic”. However, CTE is more generally associated with changes in a person’s behavior, cognitive and social abilities, making it clinically more similar to FTD. Unfortunately, CTE can only be diagnosed after death by considering the context of the patient’s history and looking at sections of their brain for abnormalities such as neuronal loss, tauopathies, as well as heightened levels of TDP-43 protein deposition. Mutations in the TDP-43 gene have also been linked to some cases of ALS, along with other neurodegenerative diseases such as FTD. Because CTE cannot be diagnosed ante mortem, it is impossible to know how many people are currently living with the disease.

There are many forms of motor neuron disorders, a few of which have been discussed here. Each varies in site of onset and the rate at which symptoms worsen – and all of them represent significant unmet medical needs when it comes to treatment. While some people find relief from some approved treatments, most do not, making the need for new treatments and cures an urgent one.
The discovery and development of effective treatments and cures for ALS is the sole mission of the ALS Therapy Development Institute. ALS TDI has several potential treatments in its research pipeline right now, including an exciting compound called AT-1501. Also, anyone diagnosed with ALS is encouraged to join the Precision Medicine Program in order to gain real time access to tools which may help them understand their own progression rate and whether or not something they are attempting treatment wise is having a desired impact.