This month, after almost five years of research by the ALS Therapy Development Institute (ALS TDI) team, ALS TDI published important findings in Nature Publishing Group’s Scientific Reports.[1] The paper introduces the possibility that aggregated clusters of SOD1 proteins, previously thought to be a disease driver of ALS, may actually protect the central nervous system (CNS) against the progression of ALS. The findings instead suggest that soluble misfolded SOD1 proteins might be the true disease driver in mutant SOD1-mediated familial ALS.
The ALS TDI study is the first to show evidence of the neuroprotective role of SOD1 protein aggregates and the toxic role of soluble misfolded proteins in the SOD1 mouse model, providing key insights into ALS research and drug development.
“The findings were surprising,” says Dr. Fernando Vieira, Chief Scientific Officer at ALS TDI. “It really took a lot of hard work by a lot of people to pull this together.” Cindy Gill invented new assays and testing methods that allowed SOD1 aggregate amounts to be compared, James Phelan spent countless hours performing brain/spinal cord dissections and measuring samples, Neurimmune shared assays and antibodies necessary to the study, and the ALS TDI in vivo team worked tirelessly to assess the neurological score and disease progression of each and every mouse model over the course of several years. The research was made possible by these contributions and the collaboration of the entire ALS TDI team.
THE RESEARCH
Scientists have long observed that abnormal clumps of protein, often called aggregates or inclusions, accumulate in certain regions of the CNS in ALS and other neurodegenerative diseases. Mutated proteins, proteins with errors in their genetic coding that cause them to misfold and aggregate, have long been identified in cases of familial ALS (FALS).[2]
Superoxide dismutase 1, commonly referred to as SOD1, is one of these mutated proteins, and is seen in about 20% of FALS cases and 2% of all ALS cases.[3] This protein is particularly important in ALS research due to the development of SOD1 transgenic mice, animal models of ALS that allow scientists to study disease progression and drug efficacy in mice with symptoms that mirror human ALS.[4]
SOD1 proteins, and proteins in general, are normally soluble. They typically undergo a complicated process in the cell by which they are correctly folded before pairing together to create a functional SOD1 dimer which is soluble in the cell’s cytoplasm, similar to how salt is dissolvable in water. In this soluble state, the SOD1 dimers move around the cell to perform their normal functions.
However, when the protein is mutated it becomes significantly more likely to misfold and as a result, it becomes more likely to aggregate. Instead of forming soluble dimer pairs, the misfolded proteins are inclined to form clusters of various sizes. Some of these clusters are small enough to remain soluble, even if misfolded. But some grow into large, insoluble aggregates greater than .2 microns – large enough to be seen with a microscope or a trained eye (see Fig. 1).
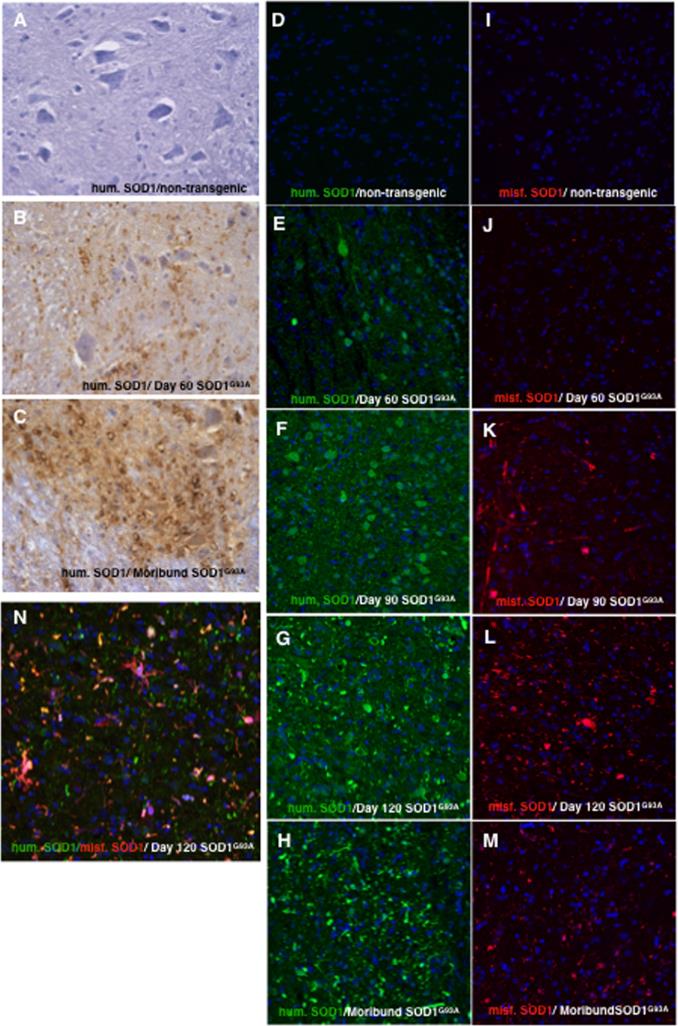
Figure 1: Immunohistochemistry and immunofluorescence of human SOD1 in lumbar spinal cord ventral horn of non-transgenic mice and in mice expressing mutant human SOD1.
The cell can handle a few of these improperly folded and clustered proteins – it tries to refold them into useful proteins or may otherwise break them down, recycling the amino acid components. But in ALS, the aggregated protein clusters may overwhelm the cell’s normal systems. The aggregates may be too large and too abundant for the cell to continuously break down and recycle all of them.
We know the malfunction in this process to be a significant factor in ALS, but the exact relationship between the symptoms of ALS seen in the SOD1 mouse model and the different variations of SOD1 (normal soluble proteins vs. soluble misfolded proteins vs. aggregated misfolded proteins – see Fig. 2) has until this point been unclear. It has been theorized that the aggregated SOD1 proteins might be clogging the cell’s axons and dendrites, disrupting essential functions within motor neurons – just as large trucks might stop the flow of traffic and cause a traffic jam.
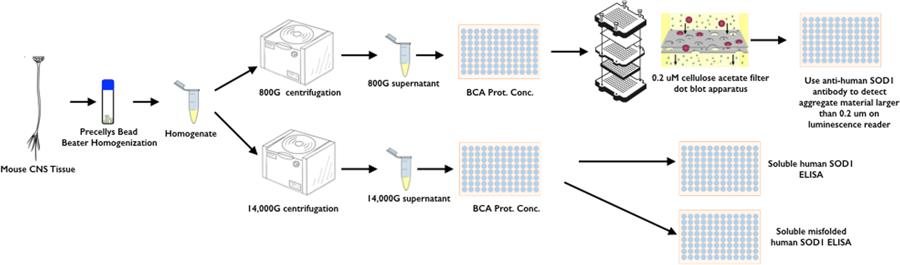 Figure 2: CNS tissue sample workflow for quantitation of total soluble human SOD1, soluble misfolded human SOD1, and SOD1 positive aggregates larger than 0.2 microns.
Figure 2: CNS tissue sample workflow for quantitation of total soluble human SOD1, soluble misfolded human SOD1, and SOD1 positive aggregates larger than 0.2 microns.
But the recent findings by ALS TDI are inconsistent with the idea that protein aggregates are causing the biggest problems in the cell. The study reveals that the regions of the CNS and spinal cord that have the most aggregated SOD1 protein are in fact the regions that are least affected by the disease. However, the regions of the CNS that have the highest proportion of soluble misfolded proteins – those misfolded proteins still small enough to be absorbed into the cytoplasm – are the regions that are most impacted by the disease and most effected by the symptoms of ALS.
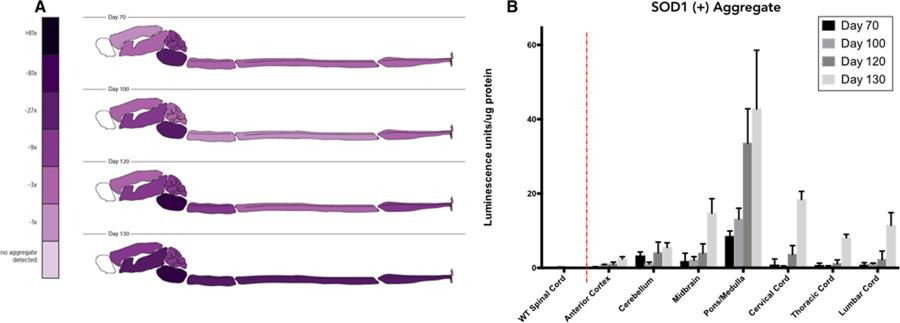
Figure 3: Human SOD1 protein aggregate in CNS of SOD1G93A mice: (A) Pictographic representation of relative SOD1 positive aggregate load in CNS (B) Bar graph of relative SOD1 positive aggregate load in 7 anatomical regions of SODG93A mouse CNS.
The findings support a newer hypothesis that “soluble misfolded SOD1 may be the disease driver in ALS,” whereas aggregated SOD1 help in the fight against the disease by allowing the cell to address its other duties and take a break in the endless process of recycling misfolded, mutated proteins.1 The aggregated proteins, once thought to be a disease driver in ALS, might in fact be neuroprotective.
THE IMPACT
SOD1 protein mutations and aggregation appear in only 20% of FALS cases and 2% of all ALS cases.3 However, aggregation of other proteins is seen in virtually 100% of ALS cases and in other neurodegenerative diseases including Alzheimer’s, Parkinson’s, and Huntington’s diseases. Any advancement in identifying the role of SOD1 protein aggregation is relevant to understanding protein aggregation more broadly and may lead to progress in the wider fields of ALS and neurodegenerative disease research.
The findings of this study suggest a de-prioritization in the development of approaches that aim to break up aggregated proteins. Drugs that attempt to dissolve aggregates have thus far been unsuccessful in achieving results. It is possible that by encouraging protein aggregation, ALS progression in SOD1 mouse models could be slowed and longevity improved, though more research is needed on this.
Additionally, the findings push for further research into the role of soluble misfolded proteins. This subset of misfolded proteins has historically been relatively difficult to detect because they are very similar to properly folded proteins. However, the recent discovery of new antibodies by biopharmaceutical company and ALS TDI-partner Neurimmune allow these soluble misfolded proteins to be more easily identified, opening up new avenues of ALS research.[5]
Find out more about ALS research at ALS TDI by joining the ALS TDI Forum, signing up for a lab tour, registering for ALS Unfiltered, an expert-led monthly webinar that dives into current research, clinical trials, and other ALS resources, or by tuning into The Endpoints Podcast, a podcast about researching, advocating for, and living with ALS.
[1] Gill, C. et al. SOD1-positive aggregate accumulation in the CNS predicts slower disease progression and increased longevity in a mutant SOD1 mouse model of ALS. Nature Scientific Reports. 9, 6724 (2019).
[2] Redler, R. L. and Dokholyan, N. V. The Complex Molecular Biology of Amyotrophic Lateral Sclerosis (ALS). Molecular Biology of Neurodegenerative Diseases. Vol. 107, 215-262 (2012).
[3] Andersen, P. M. Amyotrophic lateral sclerosis associated with mutations in the CuZn superoxide dismutase gene. Current Neurology and Neuroscience Reports. 6:37-46 (2006).
[4] Scott, S. et al. Design, power, and interpretation of studies in the standard murine model of ALS. Amyotrophic Lateral Sclerosis. 9(1):4-15 (2008).
[5] Maier, M. A human-derived antibody targets misfolded SOD1 and ameliorates motor symptoms in mouse models of amyotrophic lateral sclerosis. Science Translational Medicine. Vol. 10, Issue 470 (2018).